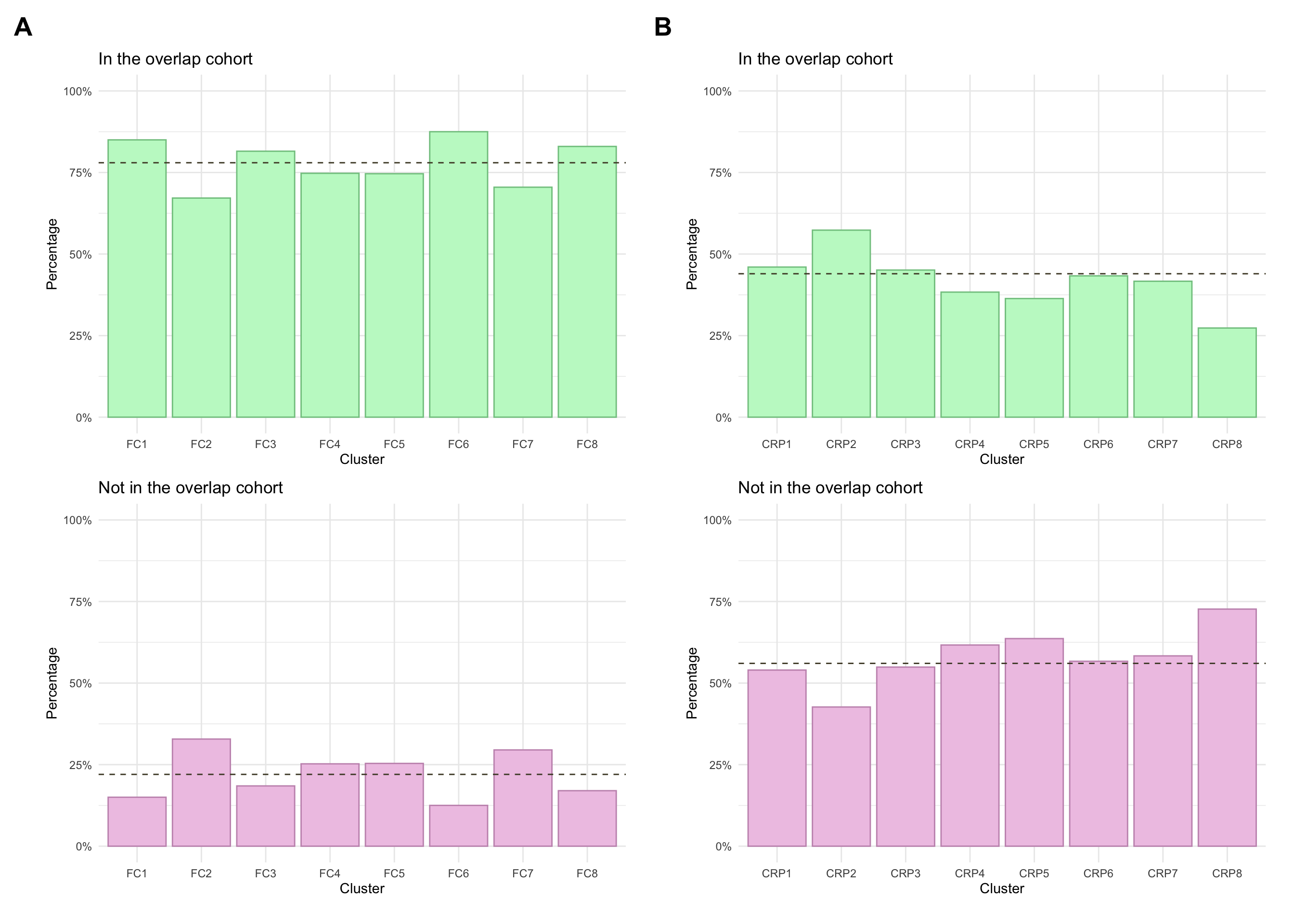
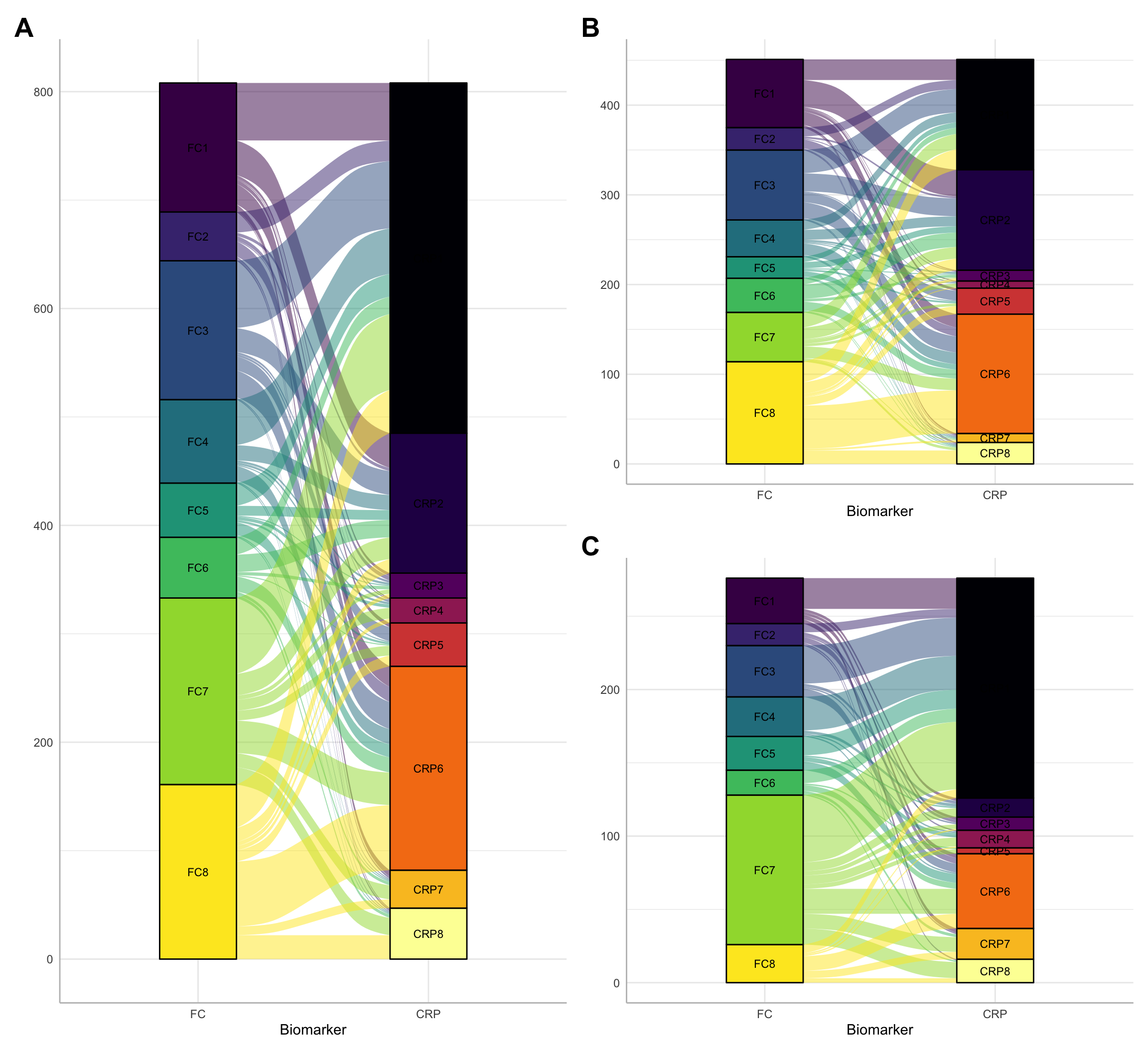
--- title: "Comparison of FC and CRP clustering" author: - name: "Nathan Constantine-Cooke" url: https://scholar.google.com/citations?user=2emHWR0AAAAJ&hl=en&oi=ao corresponding: true affiliations: - ref: CGEM - ref: HGU - name: "Karla Monterrubio-Gómez" url: https://scholar.google.com/citations?user=YmyxSXAAAAAJ&hl=en affiliations: - ref: HGU - ref: CGEM - name: "Catalina A. Vallejos" url: https://scholar.google.com/citations?user=lkdrwm0AAAAJ&hl=en&oi=ao affiliations: - ref: HGU #comments: # giscus: # repo: quarto-dev/quarto-docs --- ```{R} suppressPackageStartupMessages (library (libdr))suppressPackageStartupMessages (library (ComplexHeatmap))suppressPackageStartupMessages (library (tidyverse))library (ggalluvial)library (lcmm)library (patchwork)library (gameR)library (pander)library (libdr)set.seed (123 )<- "/Volumes/igmm/cvallejo-predicct/libdr/" ######################## ### Load FC models ### ######################## # set the number of groups <- numeric ()<- list ()<- seq (2 , 10 )for (G.cand in G.cands) {<- paste0 (prefix, "cache/fcal/ncs/fcal-" , G.cand, ".RDS" )if (file.exists (file.name)) {<- c (G.fcal, G.cand)<- readRDS (file.name)rm (G.cand, G.cands)<- readRDS (paste0 (prefix, "processed/dict.RDS" ))#################################################################### ### Load CRP moving average (no autocorrelation structure) models ### #################################################################### <- numeric ()<- list ()<- seq (2 , 10 )for (G.cand in G.cands) {<- paste0 (prefix, "cache/crp-ma/crp-" , G.cand, ".RDS" )if (file.exists (file.name)) {<- c (G.crp, G.cand)<- readRDS (file.name)rm (G.cand, G.cands)``` ```{R} #| column: page #| fig-width: 14 #| fig-height: 10 <- function (x) {:: mapvalues (x,from = seq (1 , 8 ),to = c (7 , 6 , 4 , 8 , 1 , 5 , 2 , 3 ))<- function (x) {:: mapvalues (x,from = seq (1 , 8 ),to = c (2 , 3 , 1 , 4 , 5 , 7 , 6 , 8 ))<- c (:: viridis (8 ),:: inferno (8 )<- models.fcal[[8 ]]$ pprob<- models.crp[[8 ]]$ pprob<- fcal.pprob$ ids<- crp.pprob$ ids# Only IDs in both FCAL and CRP models <- fcal.ids[fcal.ids %in% crp.ids]<- ids.comb[order (ids.comb)]<- data.frame (fcal.pprob[, c (1 , 2 )], type = "FC" )$ class <- fc.remapping (fc.classes$ class)$ class <- as.factor (paste0 ("FC" , fc.classes$ class))<- data.frame (crp.pprob[, c (1 , 2 )], type = "CRP" )$ class <- crp.remapping (crp.classes$ class)$ class <- as.factor (paste0 ("CRP" , crp.classes$ class))$ overlap <- ifelse (fc.classes$ ids %in% ids.comb, TRUE , FALSE )$ overlap <- ifelse (crp.classes$ ids %in% ids.comb, TRUE , FALSE )<- fc.classes %>% mutate (overlap = factor (levels = c (TRUE , FALSE ),labels = c ("In the overlap cohort" , "Not in the overlap cohort" ))) %>% plotCat ("overlap" , class = "class" )<- crp.classes %>% mutate (overlap = factor (levels = c (TRUE , FALSE ),labels = c ("In the overlap cohort" , "Not in the overlap cohort" ))) %>% plotCat ("overlap" , class = "class" )<- wrap_elements (p_overlap_fc) + wrap_elements (p_overlap_crp) + plot_annotation (tag_levels = "A" ) & theme (plot.tag = element_text (size = 20 , face = "bold" ))ggsave ("plots/overlap.pdf" , p, width = 14 , height = 10 )ggsave ("plots/overlap.png" , p, width = 14 , height = 10 )``` ```{R} #| column: page #| fig-width: 12 #| fig-height: 11 <- table (crp.classes$ class, crp.classes$ overlap)# Numbers for the text 8 ,2 ] / (aux[8 ,1 ] + aux[8 ,2 ])sum (aux[1 : 7 ,2 ]) / sum (aux[1 : 7 ,1 ] + aux[1 : 7 ,2 ])<- rbind (fc.classes, crp.classes)$ class <- as.factor (classes$ class)$ type <- as.factor (classes$ type)$ type <- relevel (classes$ type, "FC" )<- merge (classes,c ("ids" , "diagnosis" )],by = "ids" , all.x = TRUE ,all.y = FALSE )<- classes %>% subset (overlap == TRUE ) %>% ggplot (aes (x = type,stratum = class,alluvium = ids,fill = class,label = class+ geom_flow () + geom_stratum (alpha = 1 ) + geom_text (stat = "stratum" , size = 3 ) + theme_minimal () + theme (axis.line = element_line (colour = "gray" )) + xlab ("Biomarker" ) + scale_fill_manual (values = col.vec)<- classes %>% subset (overlap == TRUE ) %>% filter (diagnosis == "Crohn's Disease" ) %>% ggplot (aes (x = type,stratum = class,alluvium = ids,fill = class,label = class+ geom_flow () + geom_stratum (alpha = 1 ) + geom_text (stat = "stratum" , size = 3 ) + theme_minimal () + theme (axis.line = element_line (colour = "gray" )) + xlab ("Biomarker" ) + scale_fill_manual (values = col.vec)<- classes %>% subset (overlap == TRUE ) %>% filter (diagnosis == "Ulcerative Colitis" ) %>% ggplot (aes (x = type,stratum = class,alluvium = ids,fill = class,label = class+ geom_flow () + geom_stratum (alpha = 1 ) + geom_text (stat = "stratum" , size = 3 ) + theme_minimal () + theme (axis.line = element_line (colour = "gray" )) + xlab ("Biomarker" ) + scale_fill_manual (values = col.vec)<- p1 + (p2 / p3) + plot_annotation (tag_levels = "A" ) + plot_layout (guides = "collect" ) & theme (legend.position = "none" ,plot.tag = element_text (size = 20 , face = "bold" )) & guides (fill = guide_legend (nrow = 1 ))ggsave ("paper/big-comp.png" , p, width = 12 , height = 11 )ggsave ("paper/big-comp.pdf" , p, width = 12 , height = 11 )``` ```{r} <- classes.wide <- classes %>% subset (overlap == TRUE ) %>% pivot_wider (names_from = type, values_from = class)<- classes.wide %>% subset (FC == "FC1" ) %>% count (CRP) %>% mutate (percentage = (n/ sum (n) * 100 )) <- classes.wide %>% subset (CRP %in% c ("CRP1" , "CRP2" )) %>% count (FC) %>% mutate (percentage = (n/ sum (n) * 100 )) <- classes.wide %>% subset (CRP %in% c ("CRP8" )) %>% count (FC) %>% mutate (percentage = (n/ sum (n) * 100 )) <- classes.wide %>% subset (diagnosis == "Ulcerative Colitis" ) %>% subset (FC %in% c ("FC7" )) %>% count (CRP) %>% mutate (percentage = (n/ sum (n) * 100 )) ``` ```{R} #| include: false <- intersect (models.fcal[[8 ]]$ pprob$ ids, models.crp[[8 ]]$ pprob$ ids)<- subset (models.fcal[[8 ]]$ pprob, ids %in% ids.shared)[,c (1 , 2 )]colnames (fc.df) <- c ("ids" , "FCcluster" )<- subset (models.crp[[8 ]]$ pprob, ids %in% ids.shared)[,c (1 , 2 )]colnames (crp.df) <- c ("ids" , "CRPcluster" )<- merge (fc.df, crp.df, by = "ids" )<- with (mapping, table (FCcluster, CRPcluster))rowSums (tab)colSums (tab)``` ```{R} <- 0 <- 0 for (i in fc.df$ ids) {for (j in fc.df$ ids) {if (i != j) {if (subset (fc.df, ids == i)$ FCcluster == subset (fc.df, ids == j)$ FCcluster){<- paired + 1 if (subset (crp.df, ids == i)$ CRPcluster == subset (crp.df, ids == j)$ CRPcluster) {<- doublePaired + 1 / paired``` ```{R} <- 0 <- 0 for (i in crp.df$ ids) {for (j in crp.df$ ids) {if (i != j) {if (subset (crp.df, ids == i)$ CRPcluster == subset (crp.df, ids == j)$ CRPcluster){<- paired + 1 if (subset (fc.df, ids == i)$ FCcluster == subset (fc.df, ids == j)$ FCcluster) {<- doublePaired + 1 / paired``` ## Reuse {.appendix} ## {.appendix} ## Session info {.appendix} ```{R Session info} pander(sessionInfo()) ```